Institute of Technology, Resource and Energy-efficient Engineering (TREE)
Computational Chemistry Working Group
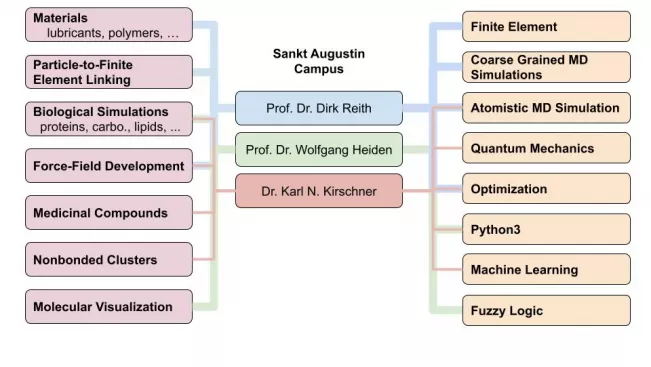
Principal Investigators
Prof Dr Dirk Reith
Prof. Dr. Wolfgang Heiden
Dr Karl N. Kirschner
Senior Researcher
Dr Marco Hülsmann
Dr. Alexander Hagg
PhD Students
Martin Schenk
Robin Strickstrock
Dirk Grommes
Jonas Bergrath
Alumni
Dr. Andreas Krämer
Dr. Christian Kandt
Roman Elfgen
Dr. Thorsten Köddermann
Research Topics and Interests
Kirschner
- Biological Modelling (Atomistic MD)
- Proteins
- Carbohydrates and polysaccharides
- Lipids
- Drug and ligand binding to proteins
- Small molecules (QM and ML)
- Conformational ensembles and elucidating potential energy surfaces
- Spectroscopy - NMR chemical shift and J-coupling; IR vibrational frequencies
- Molecular similarity (e.g. drug repurposing)
- Strengthening the interplay between ML and computational chemistry
- Nonbonded, gas-phase clusters (QM)
- Pure water clusters (e.g. (H2O)n=1-9)
- Water plus solute (e.g. Isoprene∙(H2O)n)
Reith
- Modeling and optimization of transdermal therapeutic systems
- Solubility of gases in ionic liquids
- Materials (lubricants, polymers, etc.)
- Linking particle-to-finite element methods
- Coarse graining of polymers to predict material properties for finite element simulation
Heiden
- Molecular visualization and software engineering
Kirschner & Reith
- Atomistic force-field optimization for MD simulations
- Development of software for semi-automatic force-field optimization
(COFFE = Comprehensive Optimization Force-Field Environment)
Methods
QM methods are applied on the shortest time and length scales. These methods are employed when we require high-resolution data that is reliable - at or near the level of experimental accuracy. Since QM models electron behavior, they can be used to study gas-phase chemical reactions, pathways, transition states and so on. However, they are also useful for atomistic understanding of condense-phase behavior.
Classical atomistic MD simulation provide us access to longer time and length scales. These methods are used when we need to simulate larger systems in solution. Here we can explore phase changes, the role of solvent, or the binding of a drug to a protein. Statistical sampling and analysis is heavily focused upon to ensure reproducibility and to prevent inaccurate conclusions being drawn.
This is complemented by the technique of mesoscale coarse-grained MD simulations. Coarse graining extends the time and length scale further. These models enable us to increase our statistical sampling, which is important in polymer modeling.
Data-driven ML helps us to improve the physics-based models described above, and can generate new molecule-focused hypotheses for us to explore. We can also use ML methods to perform data analysis - for example, reducing a dataset’s dimensionality (PCA, t-SNE), identifying similarity, or feature correlation.
Hardware
The computer cluster of Bonn-Rhein-Sieg University of Applied Sciences is used for computations. An efficient implementation of the method and effective parallelization enable high performance calculations that lead to accurate simulation results.
Selected publications
- Freisleben, Fabian, Franziska Modemann, Jana Muschhammer, Hauke Stamm, Franziska Brauneck, Alexander Krispien, Carsten Bokemeyer, Karl N. Kirschner, Jasmin Wellbrock, and Walter Fiedler. "Mebendazole Mediates Proteasomal Degradation of GLI Transcription Factors in Acute Myeloid Leukemia." Intern. J. Molec. Sci. 22, 10670 (2021).
- K. N. Kirschner, W. Heiden, D. Reith: "Small Alcohols Revisited: CCSD(T) Relative Potential Energies for the Minima, First-and Second-Order Saddle Points, and Torsion-Coupled Surfaces", ACS Omega 3 (1), 419-432 (2018).
- S. Knauer, M.R. Schenk, T. Köddermann, D. Reith, P. Jaeger: "Interfacial Tension and Related Properties of Ionic Liquids in CH4 and CO2 at Elevated Pressures: Experimental Data and Molecular Dynamics Simulation", J. Chem. Eng. Data 62, 2234-2243 (2017).
- K. N. Kirschner, W. Heiden, D. Reith: "Relative electronic and free energies of octane's unique conformations", Molec. Phys. 115, 1155-1165 (2017).
- M. Hülsmann, K. N. Kirschner, A. Krämer, D. D. Heinrich, O. Krämer-Fuhrmann, D. Reith: "Optimizing Molecular Models Through Force Field Parameterization via the Efficient Combination of Modular Program Packages", Foundations of Molecular Modeling and Simulation, 53-77, Springer (2016).
- A. Krämer, M. Hülsmann, T. Köddermann, D. Reith: "Automated parameterization of intermolecular pair potentials using global optimization techniques", Computer Physics Communications 185, 3228-3239 (2014).
Partners and Collaborators
- Department of Chemical Engineering and Materials Science, University of California, Davis, CA, USA (Prof. Dr. Roland Faller)
- Department of Chemistry, Furman University, Greenville, SC, USA (Prof. Dr. George Shields)
- Institute for Integrated Natural Sciences, University of Koblenz, Koblenz, Germany (Prof. Dr. Wolfgang Imhof)
- Institute of Process Engineering, Technical University of Berlin, Berlin, Germany (Prof. Dr. Jadran Vrabec)
- Institute for Simulation Technology and Scientific Computing, University of Siegen, Siegen, Germany (Prof. Dr.-Ing. Sabine Roller)
- Max Planck Institute for Polymer Research, Mainz, Germany (Prof. Dr. Kurt Kremer, Dr. Torsten Stühn)