Institut für Technik, Ressourcenschonung und Energieeffizienz (TREE)
Arbeitsgruppe Computational Chemistry
In diesem Kontext legt die Forschungsgruppe „Computational Chemistry“ den Schwerpunkt auf molekulardynamische Simulationen zur Untersuchung der sogenannten “weichen Materie” (z.B. Kunststoffe, Schmiermittel, Ionische Flüssigkeiten oder biochemische Substanzen). Im Zentrum der Aktivitäten stehen seit langem zwei Bereiche: zum einen die Entwicklung und Optimierung von Kraftfeldern mit eigenentwickelten Open Source Werkzeugen. Dies wird benötigt, um sicherzustellen, dass die Modelle die Realität so gut wie möglich abbilden. Zum anderen ist die Bearbeitung von Aufgabenstellungen, die aus der praktischen ingenieurwissenschaftlichen Anwendung getrieben sind, wesentliches Kennzeichen der Forschungsgruppe, um z.B. Beiträge zur Material- bis hin zur Wirkstoffentwicklung machen zu können. Neu hinzugekommen ist die Verbindung zur “Virtual Reality”, d.h. hier konkret die geschickte Visualisierung wissenschaftlicher Daten, um auf einer weiteren Ebene für Erkenntnisgewinn zu sorgen.
„Creating and using models have a long history in scientific research and education. Computational chemistry is an exciting field that requires cross-disciplinary knowledge to build realistic and trustworthy models. Because of this, one always has the opportunity to learn something new, apply ideas from one field to others, and create new knowledge at incredible resolution levels.”
Dr. Karl Kirschner

Aktuelle Forschungsthemen
- Entwicklung eines Software-Pakets zur semi-automatischen globalen und lokalen Optimierung von Kraftfeldern (FFLOW = Force FieLd Optimization Workflow)
- Optimierung intramolekularer Kraftfeldparameter mittels quantenmechanischer Berechnungen
- Modellierung und Optimierung transdermaler therapeutischer Systeme
- Untersuchung der Löslichkeit von Gasen in ionischen Flüssigkeiten
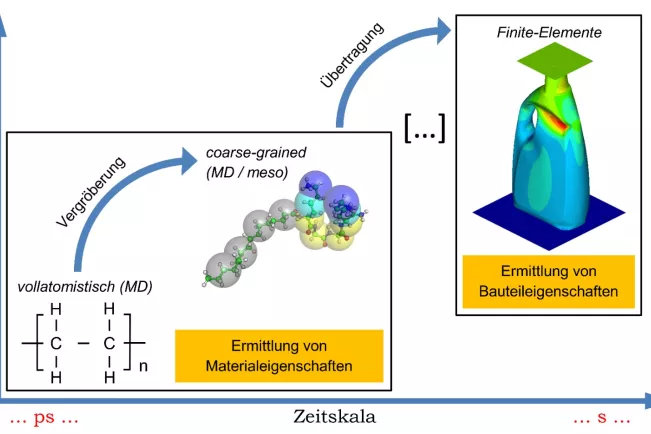
- Coarse Graining von Polymeren zur Ermittlung von Materialkennwerten für die Finite-Elemente-Simulation
- Simulationen der Bindung biologisch aktiver Moleküle an krankheitsassoziierte Proteine (z.B. im Kontext Akuter Myeloischer Leukämie (AML), Myopathien, oder Krebs).
Methoden, Kompetenzen und Anwendungen
Im Sinne eines Vorgehens auf mehreren Betrachtungsebenen decken die Aktivitäten der Forschungsgruppe unterschiedliche Zeiträume und Größenordnungen ab. Zum Einsatz kommen quantenmechanische Methoden, die beispielsweise zur Optimierung von Kraftfeldern für die klassische molekulardynamische Simulation (MD) notwendig sind. Zur Optimierung werden unterschiedliche lokale und globale Strategien, auch unter Hilfestellung von maschinellem Lernen, eingesetzt. Die (voll-)atomistische MD-Simulation bildet den Kern der in der Forschungsgruppe eingesetzten Methoden. Sie erlaubt die Bearbeitung der überwiegenden Zahl an Problemstellungen. Zur Abbildung von Effekten auf längeren Zeitskalen findet zudem die Technik des Mesoscale Coarse Graining (CG) Anwendung. Coarse Graining-Methoden sind von besonderer Attraktivität, da sie es ermöglichen, sowohl die simulierte Zeit als auch die Größe der untersuchten Systeme wesentlich zu erhöhen. Hierzu wird das atomistisch vollständig aufgelöste System in ein vergröbertes, mesoskopisches System überführt, indem Atomgruppen zu „Superatomen“ zusammengefasst werden.
Aus der Bandbreite an unterschiedlich auflösenden Modellansätzen folgt eine Vielzahl an Systemen und Anwendungsfeldern:
- Gase und Flüssigkeiten mit niedrigem Molekulargewicht
- Ionische Flüssigkeiten
- Polymere in der Schmelze
- Biologische Systeme, die Proteine, Lipide, Kohlenhydrate und Lignin umfassen
- weitere Anwendungen in herausfordernden industriellen Problemstellungen
In Ergänzung zur Anwendung von Methoden und Simulationstools steht zusätzlich die Weiterentwicklung derselben im Fokus. Arbeiten der Gruppe befassen sich u.a. mit folgenden Punkten:
- Entwicklung eines Workflows für die (semi-)automatische Kraftfeldentwicklung von chemischen Verbindungen
- Automatisierte Optimierung von Kraftfeldern in der molekularen Modellierung
- Entwicklung neuer Methoden und Werkzeuge für Simulationen "weicher Materie"
Labore

Die Berechnungen werden auf dem leistungsfähigen Rechencluster der Hochschule Bonn-Rhein-Sieg durchgeführt. Möglichkeiten einer effizienten Parallelisierung in Verbindung mit Simulationswerkzeugen auf dem Stand der Wissenschaft erlauben detailgenaue Berechnungen.
Lehre

Im Rahmen des Forschungsprojekts UMMBAS arbeitet die Gruppe derzeit (2023-25) an der Entwicklung einer kollaborativen VR-Umgebung, um über Standortgrenzen hinweg effizient an Themen aus dem Bereich Molecular Modelling zusammenarbeiten zu können. Sind die zu entwickelnden Kollaborationskonzepte erst einmal etabliert, werden sie absehbar auch in der Lehre einsetzbar sein. Dabei könnten sie insbesondere in fachbereichs- oder hoschschulübergreifend angebotenen Lehrmodulen zum Einsatz kommen.










Kooperationspartner
- Edward E. Whitacre Jr. College of Engineering, Texas Tech University (Prof. Dr. Roland Faller)
- Institut für Integrierte Naturwissenschaften, Universität Koblenz (Prof. Dr. Wolfgang Imhof)
- Institut für Prozess- und Verfahrenstechnik, Technische Universität Berlin (Prof. Dr. Jadran Vrabec)
- Department of Chemistry, Furman University, South Carolina (Prof. Dr. George Shields)
- Division of Hematology, Ohio State University, Comprehensive Cancer Center, Columbus, OH USA (Prof. Drs. John Byrd and Karilyn Larkin)
Ausgewählte Publikationen
Arbeitsgruppe Computational Chemistry
2023
Alexander Hagg, Karl N. Kirschner:
Open-Source Machine Learning in Computational Chemistry.
PDF Download (CC BY-NC-ND 4.0) doi:10.1021/acs.jcim.3c00643 PMID urn:nbn:de:hbz:1044-opus-74406
BibTeX | RIS
Max Müller, Alexander Hagg, Robin Strickstrock, Marco Hülsmann, Alexander Asteroth, Karl N. Kirschner, Dirk Reith:
Determining Lennard-Jones Parameters Using Multiscale Target Data through Presampling-Enhanced, Surrogate-Assisted Global Optimization.
doi:10.1021/acs.jcim.2c01231 PMID
BibTeX | RIS
JinHeng Lin, Sean M. Gettings, Khaoula Talbi, Rainer Schreiber, Michael J. Taggart, Matthias Preller, Karl Kunzelmann, Mike Althaus, Michael A. Gray:
Pharmacological inhibitors of the cystic fibrosis transmembrane conductance regulator exert off-target effects on epithelial cation channels.
PDF Download (CC BY 4.0) doi:10.1007/s00424-022-02758-9 PMID urn:nbn:de:hbz:1044-opus-64572
BibTeX | RIS
2022
Dirk Grommes, Martin R. Schenk, Olaf Bruch, Dirk Reith:
Initial Crystallization Effects in Coarse-Grained Polyethylene Systems After Uni- and Biaxial Stretching in Blow-Molding Cooling Scenarios.
PDF Download (CC BY 4.0) doi:10.3390/polym14235144 PMID urn:nbn:de:hbz:1044-opus-65219
BibTeX | RIS
Walter Fiedler, Fabian Freisleben, Jasmin Wellbrock, Karl N. Kirschner:
Mebendazole's Conformational Space and Its Predicted Binding to Human Heat-Shock Protein 90.
doi:10.1021/acs.jcim.2c00290 PMID
BibTeX | RIS
Robin Strickstrock, Marco Hülsmann, Dirk Reith, Karl N. Kirschner:
Optimizing Lennard-Jones parameters by coupling single molecule and ensemble target data.
doi:10.1016/j.cpc.2022.108285
BibTeX | RIS
2020
Meera C. Viswanathan, William Schmidt, Peter Franz, Michael J. Rynkiewicz, Christopher S. Newhard, Aditi Madan, William Lehman, Douglas M. Swank, Matthias Preller, Anthony Cammarato:
A role for actin flexibility in thin filament-mediated contractile regulation and myopathy.
doi:10.1038/s41467-020-15922-5 PMID
BibTeX | RIS
Wiebke Ewert, Peter Franz, Georgios Tsiavaliaris, Matthias Preller:
Structural and Computational Insights into a Blebbistatin-Bound Myosin•ADP Complex with Characteristics of an ADP-Release Conformation along the Two-Step Myosin Power Stoke.
PDF Download (CC BY 4.0) doi:10.3390/ijms21197417 urn:nbn:de:hbz:1044-opus-50854 PMID
BibTeX | RIS
Karl N. Kirschner, Dirk Reith, Wolfgang Heiden:
The performance of Dunning, Jensen, and Karlsruhe basis sets on computing relative energies and geometries.
doi:10.1080/1539445x.2020.1714656
BibTeX | RIS
Martin R. Schenk, Thorsten Köddermann, Karl N. Kirschner, Sandra Knauer, Dirk Reith:
Molecular Dynamics in the Energy Sector: Experiment and Modeling of the CO2/CH4 Mixture.
doi:10.1021/acs.jced.9b00503
BibTeX | RIS
2019
Austen Bernardi, Roland Faller, Dirk Reith, Karl N. Kirschner:
ACPYPE update for nonuniform 1–4 scale factors: Conversion of the GLYCAM06 force field from AMBER to GROMACS.
doi:10.1016/j.softx.2019.100241
BibTeX | RIS
Kontakt

Dirk Reith
Mathematik, Physik und Simulationsanwendungen (Forschungsprofessur), Direktor des TREE-Instituts, Faculty Advisor BRS Motorsport (Formula Student)
Forschungsfelder
Standort
Sankt Augustin
Raum
B 223
Adresse
Grantham-Allee 20
53757 Sankt Augustin
Telefon
+49 2241 865 9678